

I recently published an article questioning the idea that a lifter’s p-ratio will inherently get worse as body-fat percentage increases. More specifically, I pointed out that a p-ratio is really just a descriptive summary of two relatively independent processes (changes in fat mass and changes in lean mass), and pointed out that all arguments pointing at “impaired p-ratios” at high body-fat levels are really just implying blunted hypertrophy at high body-fat levels. In my article, I described why the case for impaired hypertrophy at higher body-fat levels is very weak, using several different lines of evidence. If you’re unfamiliar with p-ratios or the conversation surrounding them, you’ll definitely want to check out that article (or at least the summary) before diving into this one.
I spent some time pointing out that the most common mechanism “blamed” for poorer p-ratios at higher body-fat levels (impaired insulin sensitivity) lacked plausibility, but the more important point (by far) was that no one has presented any compelling evidence that a problem actually exists. In other words, despite laborious theorizing about underlying mechanisms, no one seems to have their hands on any data indicating that lifters with high body-fat actually struggle to achieve hypertrophy or make lean gains in comparison to leaner lifters. The evidence-based fitness community has largely embraced this p-ratio concept based on a shaky “cause,” but has failed to empirically establish the existence of the actual “effect” that the cause is purported to elicit.
Shortly after my article was published, someone named Menno Henselmans published a rebuttal. In the rebuttal, Menno largely shifts the blame from insulin sensitivity to inflammation; the purported “cause” is shifted, but it brings us no closer to evidence of the actual “effect” it’s intended to explain. Menno’s rebuttal provides no additional data to indicate that p-ratios or hypertrophy are impaired at higher body-fat levels in lifters. It also implies an alternative model by which inflammation, and potentially a worsened hormonal environment, impairs p-ratios as body-fat increases, but the model has some gaps and internal contradictions. While I tackled the original p-ratio article solo (with Greg assisting with some data aggregation), we have a lot to cover here, so Greg and I are teaming up on this rebuttal. As a result, you’ll see a mixture of singular (I/me) and plural (us/we) language. In this article, we will respond to the criticisms in Menno’s rebuttal, highlight the gaps and contradictions in the alternative model, and reiterate the general lack of evidence to substantiate this popular p-ratio hypothesis.
Topic: Forbes and Hall Models
Menno and I generally agree that there are major limitations when trying to apply the Forbes and Hall models to people who are doing resistance training. However, Menno makes two statements I disagree with:
“…if fat related factors in our body affect nutrient partitioning measurably even in sedentary individuals, you could also argue that these effects could be even more pronounced in strength trainees, where the potential for muscle growth is of course far greater.”
Menno will later commit to a couple of proposed mechanisms. He suggests that chronic inflammation is one of the primary reasons why people with high body-fat would have worse p-ratios. However, this mechanism should be more present in sedentary individuals, and less notable in people that are training. As noted in a review by You et al:
Current evidence supports that exercise training, such as aerobic and resistance exercise, reduces chronic inflammation, especially in obese individuals with high levels of inflammatory biomarkers undergoing a longer-term intervention.
Menno also states:
“…The relation goes both ways: leaner individuals are also at greater risk of muscle loss. However, this is where strength training could make a crucial difference. Sedentary individuals will almost always lose both muscle and fat in energy deficit (typically 25% of weight loss is lean mass) while gaining both muscle and fat in energy surplus (mostly fat of course). However, this is no longer the case in strength trainees.”
As noted in my original article, I certainly agree that the Forbes and Hall models work in both directions, and indicate that leaner people lose more lean mass during weight loss. I also agree that resistance training would undoubtedly modify this relationship. However, this statement seems to imply that resistance training would specifically influence p-ratios during weight loss, so it’s important to reinforce that resistance training would certainly influence the “other” direction as well (that is, drastically alter the lean mass gained during weight gain). For this reason, I think the Forbes and Hall models are fully unusable for our purposes. I don’t think Menno necessarily disagrees with that, but I wanted to highlight these two comments to make sure readers don’t warm back up to the idea that these Forbes and Hall models can be applied to longitudinal resistance training applications in either direction (weight loss or weight gain).
Topic: The Rodent Study by De Sousa et al (2020)
Menno disagrees with my interpretation of this study. First, he states:
“The diets did not in fact promote similar degrees of obesity. The high-fat diet induced a 60% increase in bodyweight, significantly greater than the 36% in the Western group.”
You can see the raw weight gain values here:
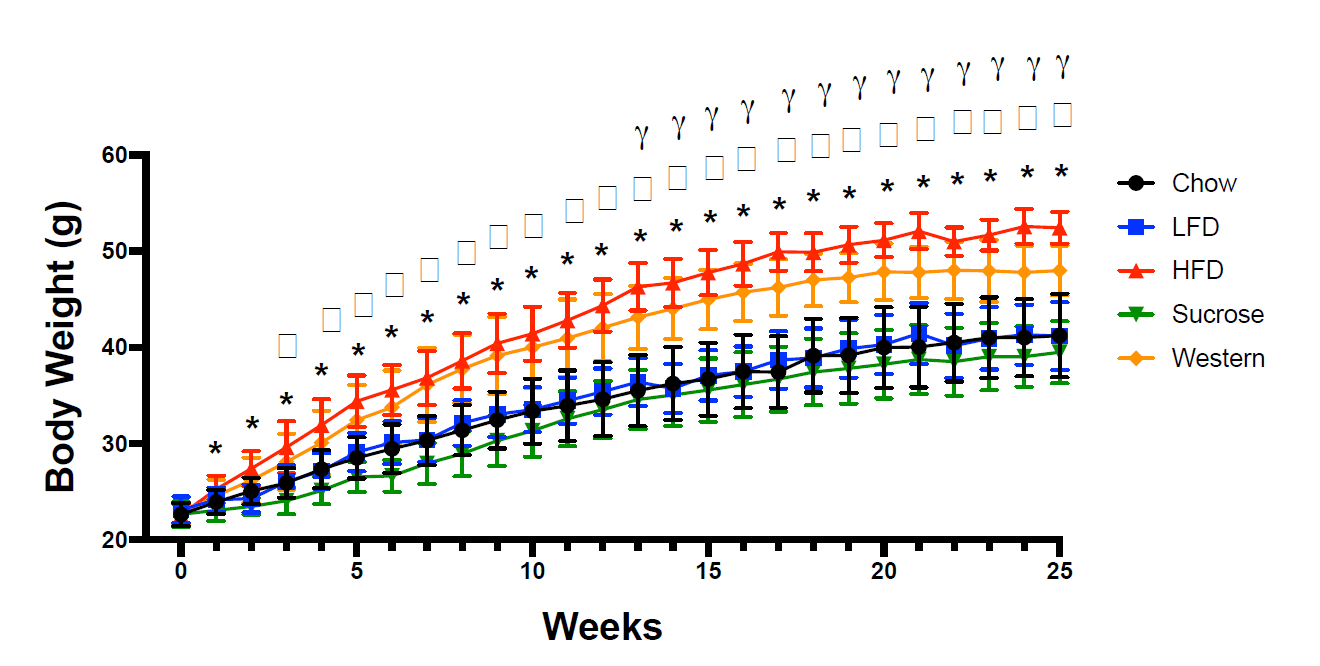
Over the course of the 25-week feeding period, both the high-fat diet and Western diet groups started around 22g, and ended up well above 40g. Weight gain differed a little bit, with more weight gain in the high-fat diet group, but both groups approximately doubled their body mass, and both gained noticeably more weight than the other groups. The researchers report that the magnitude of weight gain was significantly different between the high-fat and Western groups, and that the high-fat group gained 60% more than the standard chow group and the Western group gaining 36% more than the standard chow group. However, today is not the day that I begin treating total body mass as a better indicator of obesity status than body-fat percentage. If you look at the body-fat percentage data, you see that both groups had much higher body-fat than the other three groups after the weight gain phase of the study:
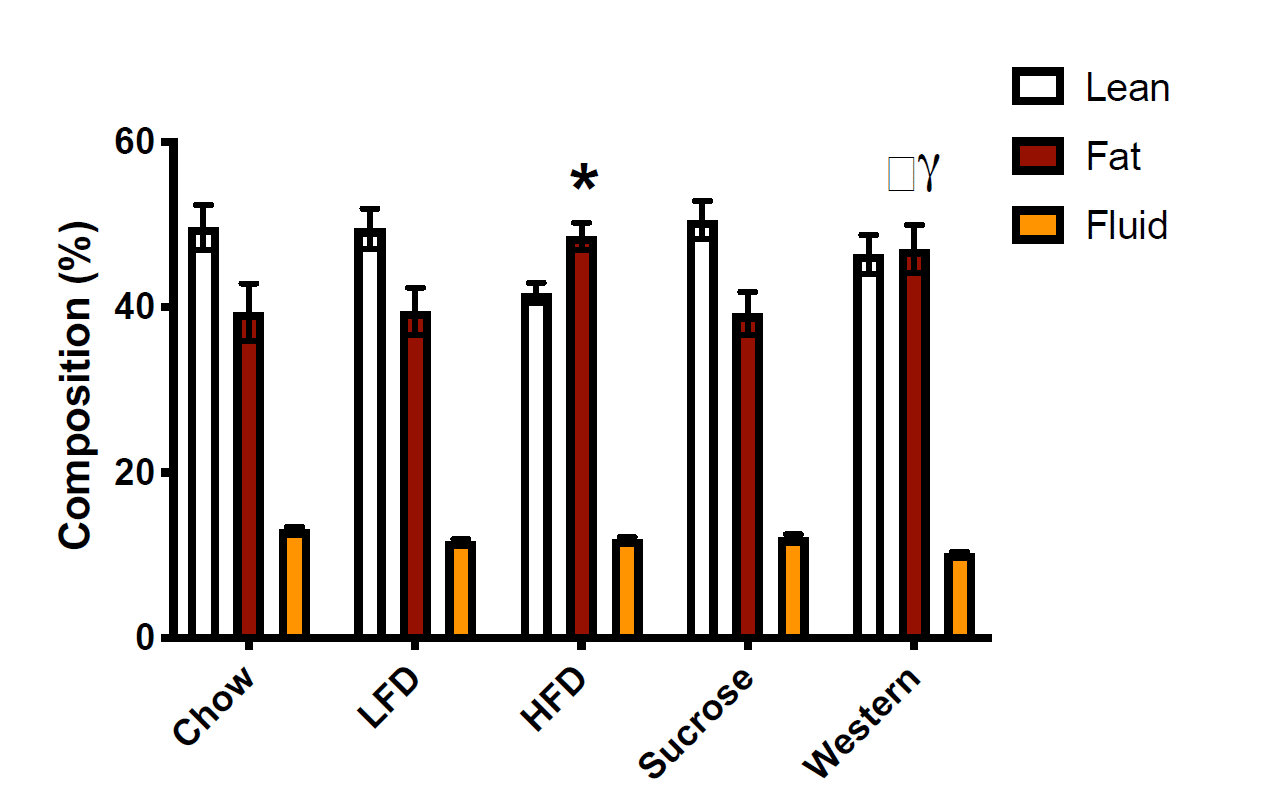
I extracted values from this figure using WebPlotDigitizer. You may get slightly different values depending on how exact you are with the extraction process, but my extraction indicated that body-fat percentages were all between 38.8-39.1% in the chow, low-fat, and sucrose groups. In contrast, the high-fat diet group was about 48.1% body-fat, and the Western diet group was about 46.6% body-fat. As you’d expect, the high-fat and Western groups both had significantly higher body-fat percentages than all other groups. The researchers seem to report that the difference between the high-fat and Western diets is significant, but I don’t see how that’s the case. If you extract the means and standard deviations and compare the two groups using a t-test, the body-fat values in the high-fat and Western diets are not significantly different from one another. If I had to guess, I’d speculate that they actually ran the stats on the raw values and found that absolute fat mass was higher in the high-fat group than the Western group, then decided to present the data as percentages after the fact, but that’s speculative. The important point is that the high-fat diet group and Western group had similar body-fat percentages, and they were both markedly higher than the three other groups, which were similar to one another. Obesity was successfully induced in both groups, and they were not meaningfully (or significantly) different from one another in terms of body-fat percentage.
Menno also states:
“…Overall, it seems that the fatter the mice got, the worse their subsequent gains. I’d say this study is actually strong proof of principle that getting too fat can reduce your gains.”
This statement implies a dose-response relationship, which was not observed. As noted above, there was a clear divergence in body-fat levels: the high-fat and Western groups had markedly higher body-fat percentages than the other groups, with only a modest difference between them. Here are the hypertrophy values after the entire 30-day loading period:
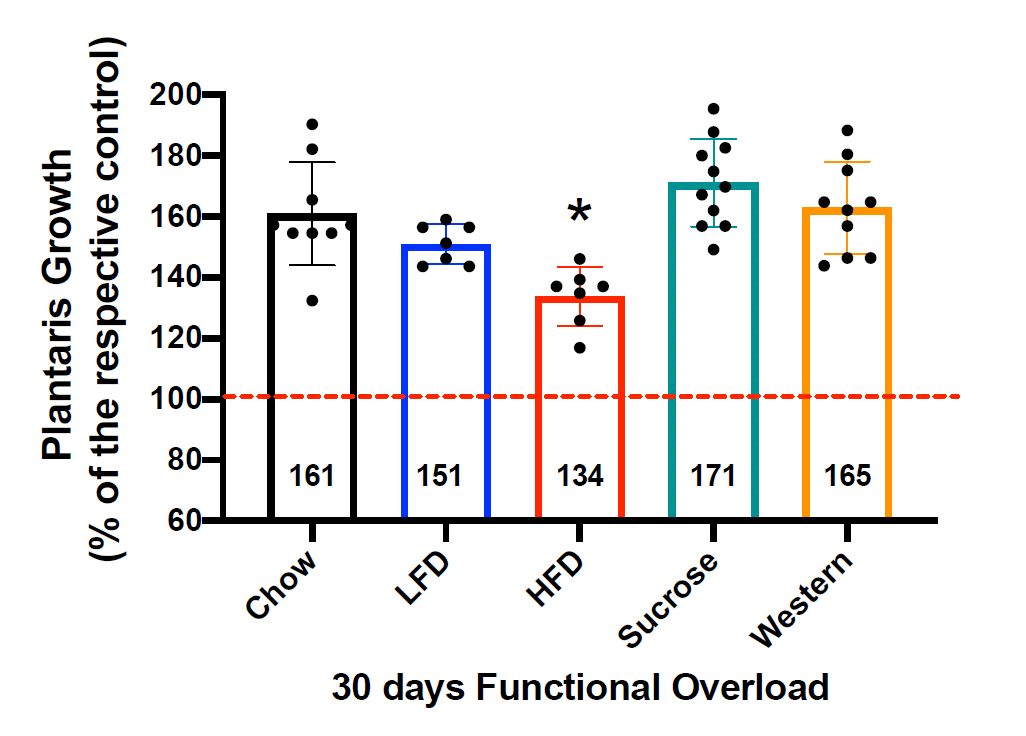
It is exceedingly difficult to suggest that this reinforces a dose-response relationship between body-fat level and observed hypertrophy. If anything, it suggests a relationship in which body-fat has absolutely no effect on hypertrophy whatsoever until you get to a magic threshold that exists, somewhere between precisely 46.6% and 48.1% body-fat. The Western group, which got to about 46.6% body-fat, had the second-highest relative hypertrophy values, whereas hypertrophy was markedly lower in the high-fat diet group, which got to about 48.1% body-fat. Personally, I think the more suitable interpretation is that the difference in diets was more impactful than the <2% gap in body-fat percentage, as do the authors of the study.
Topic: Insulin Sensitivity
Overall, Menno generally seems to agree that there’s minimal justification for the idea that higher body-fat impairs gains through a mechanism related to insulin sensitivity, so there isn’t much to discuss here.
Topic: Inflammation
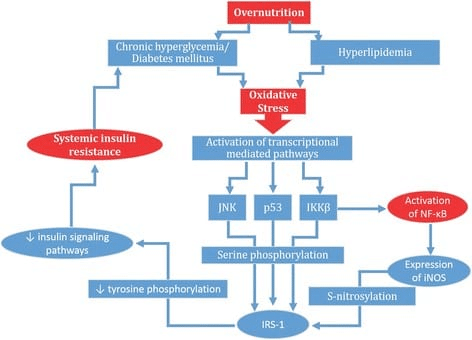
In Menno’s rebuttal, he claims that I missed an important body of literature indicating that high body-fat levels impair gains due to higher levels of chronic inflammation. In fact, I would argue that we addressed the interrelationship between higher body-fat, inflammation, and hypertrophy similarly well, insofar as we each cited an equal number of studies documenting impaired muscle hypertrophy due to inflammation induced by high body-fat percentage in resistance trained subjects (zero). Menno explains the mechanistic basis for his assertion that inflammation blunts hypertrophy in lifters with higher body-fat percentages, which is pretty concisely reflected in a figure that he included from this article:
The overarching theme of my initial article was that we could theorize our way to a nearly infinite number of plausible effects that don’t actually pan out in real life. I am far more interested in obtaining evidence of an effect before speculating about which cause is most defensible; until we can empirically establish that gains are impaired at high body-fat levels, purported causes are inconsequential. It’s also worth noting how the section about insulin sensitivity was framed in our initial article: “As a reminder, the primary mechanism most commonly used to support this hypothesized relationship between higher body-fat and impaired hypertrophy is that excess fat mass leads to insulin resistance in muscle cells, which shunts energy and nutrients toward fat mass and away from muscle cells, thereby favoring fat gain and attenuating muscle growth.” It was never suggested that this is the only potential mechanism people have kicked around; it’s just the mechanism we see discussed most often. In the interest of keeping an article from becoming a book, we decided to focus on the most commonly discussed mechanism, rather than addressing all plausible mechanisms. We chose not to address every potential mechanism, because we believe longitudinal data assessing the actual outcome we care about (changes in lean mass versus changes in fat mass in people with different levels of body-fat) is far more relevant.
In terms of empirical support for this idea that high body-fat impairs hypertrophy via inflammation, Menno links to one of his previous articles on inflammation and muscle growth. If obesity is impairing gains through inflammation, and this is the link Menno is using to solidify his point, we should justifiably expect to see that the article will lead us toward studies indicating that body-fat is an important factor linking inflammation to impaired gains. Menno’s inflammation article cites four main articles to support this point.
Based on the first of the studies, he suggests that “non-responders to strength training, individuals that don’t make any gains from a training program, have been found to have increased inflammation (TNF-alpha) levels.” The study was carried out in untrained postmenopausal women who were over 60 years old. Interestingly, the non-responders purportedly being held back by inflammation were slightly leaner than the responders at baseline (41.5% vs. 43.1% body-fat), so baseline body-fat wasn’t nearly impactful enough to steer the eventual results. Also, the study measured two other inflammation biomarkers (CPR and IL-6), which had no statistically significant impact on outcomes. The study also found no significant correlations between any of their inflammation biomarkers and muscle mass or strength. So, this study linked one of three inflammation markers to gains in lean mass, but baseline body-fat was measured and had absolutely nothing to do with the relationship, suggesting that it was completely overshadowed by more impactful factors driving inflammation.
Based on a second study, he suggests that “In a 9 month study on postmenopausal, strength training women, trunk fat mass correlated with resting IL-6 levels and negatively correlated with muscle growth.” This study was again carried out in postmenopausal women, with an age range of 44-69 years old. More specifically, changes in muscle mass were negatively correlated with the change in IL-6 (not baseline IL-6), and changes in IL-6 were positively correlated with changes in trunk fat (but not changes in total body-fat, which directly contradicts this idea that body-fat percentage was driving the observed effects). It’s important to note that this was a nine-month study with high ecological validity, but a low degree of control. The training intervention was “at least” two workouts per week, and the results don’t give us any indication about whether or not the number of workouts people chose to do, or their adherence to the program in general, impacted results. Nonetheless, one might want to theoretically connect these results to body-fat, given that muscle growth was negatively correlated with changes in IL-6, and changes in IL-6 were positively correlated with changes in trunk fat. However, the researchers divided up the participants into two groups: one group (n = 11) gained ≥5% trunk fat during the study and experienced an increase in IL-6, and the other group (n = 11) gained <5% trunk fat and did not experience an increase in IL-6. Over nine months of lifting, these previously untrained lifters gained only 0.9kg of muscle mass in the lower-inflammation group, and lost 0.6kg of muscle mass in the higher-inflammation group. So, did this high-inflammation group fail because they had high body-fat at baseline? Nope; their body-fat percentage was slightly lower at baseline (40.8% versus 41.7%). Did they fail because gains in total body-fat drove their IL-6 levels upward? Nope; the researchers reported that increases in IL-6 were correlated with changes in abdominal fat, but not with changes in total body-fat.
In my article about p-ratios, the question was largely aimed at answering two questions:
- Can someone with high body-fat make lean gains?
- Would someone necessarily improve their ability to make lean gains by losing fat before their muscle gaining phase?
The results of this study tell us that your body-fat level going into the muscle gaining phase has very little to do with outcomes, while large increases in systemic inflammation (or things that increase inflammation) during the intervention completely supersede the importance of the body-fat level you started at. So, why does a group of untrained people lose muscle mass over nine months of lifting? I’m not sure. It could relate to adherence, which we have no information about. It could be that they did all the stuff we know to be independently bad for gains (high stress, poor sleep, excessive sedentary time, crappy diet), which also happen to elevate systemic inflammation. It’s also possible that the observations in this study are specific to post-menopausal women.
I hate when people assume poor generalizability without any basis for their assumption, but there is justification in this case. In the study cited by Menno, the authors discussed the fact that women tend to accumulate visceral fat during menopause via hormone-dependent mechanisms, and it’s important to note that visceral fat (which would be reflected by “trunk fat” in their study) releases more IL-6 per gram of tissue than subcutaneous fat. Changes in IL-6 were correlated with changes in trunk fat, but not with changes in total body-fat, and time of menopause was inversely associated with changes in muscle mass. The authors also reported that time of menopause was correlated with changes in IL-6; the magnitude of this relationship is unclear, but it lines up well with other studies showing that IL-6 values tend to increase over time in post-menopausal women with or without exercise (although exercise may attenuate that increase to some extent). With these factors in mind, it’s quite possible that this study’s results were meaningfully impacted by proximity to the onset of menopause, which could have contributed to visceral fat accumulation and higher IL-6 levels. It’s also possible that the results could have been meaningfully impacted by any variety of inflammation-promoting habits that were not assessed or controlled. But to reiterate, the results weren’t driven by baseline differences in total body-fat or changes in total body-fat.
Based on a third study, he suggests that “Elevated pro-inflammatory gene expression has also been found to occur during overreaching and was associated with reduced muscle growth compared to training with less volume.” This study was also carried out in older adults (aged 60-75 years old); the sample was roughly half male and half female, and all female participants were postmenopausal. The study involved four training groups with varying degrees of weekly training frequency and intensity. Training programs involved harder high-resistance days (H) and easier low-resistance days (L); two groups did two weekly training sessions (HL and HH) while the other two did three weekly training sessions (HLH and HHH). The premise here is that the second-hardest program (HLH) induced the best gains, but the hardest program (HHH) induced overreaching, which increased inflammation, which led to worse gains than the HLH group. I would push back against that premise. Compared to all other groups, the HLH group had the best results in terms of total lean mass gains and thigh muscle mass gains. For dynamic lower-body strength outcomes, the HLH group made larger improvements than the HHH group for knee extension 1RM (HHH +26% versus HLH +36%) and equivalent gains in leg press 1RM (HHH +29% versus HLH +29%). The HLH group also made better gains in isometric leg extension force, but I’m not super interested in isometric gains from these entirely dynamic training programs. The strength results for leg press and knee extension 1RM are presented in the figures below:
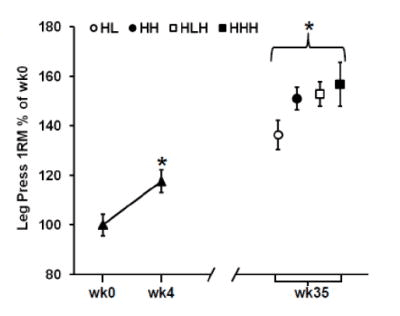
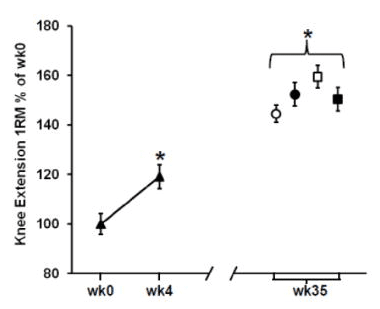
In contrast, the HHH group had the largest gains in arm muscle mass compared to all other groups (a little over 400g, versus <300g for all other groups). Further, the HHH group had larger increases than the HLH group for chest press 1RM (HHH +25% versus HLH +16%) and arm curl 1RM (HHH +55% versus HLH +36%), with identical increases in overhead press 1RM (HHH +14% versus HLH +14%). Here are the figures presenting the results for arm muscle mass and arm curl 1RM:
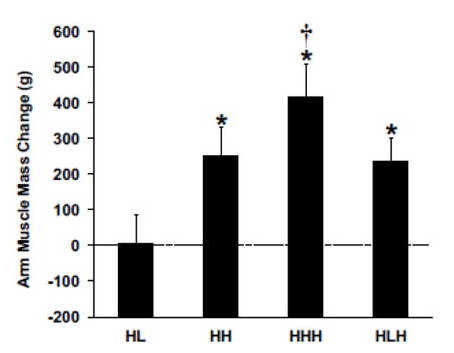
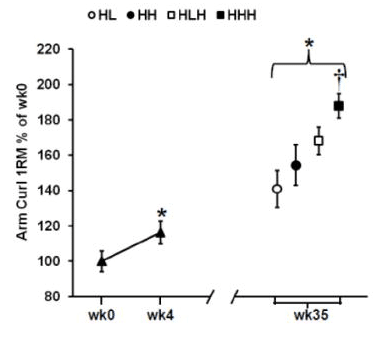
It seems hard to say that the HHH group was having all their gains ruined by systemic inflammation, given that they got plenty big and plenty strong (experiencing the largest or second-largest gains in most of the outcome measures), and the impairments seemed to be muscle group specific. It also seems hard to make any inferences about systemic inflammation in this study, since what the researchers actually assessed was the local expression of cytokine receptors in the quads.
This study might say something about overtraining, or it might say something about training variables and diminishing returns, but most importantly, it has literally nothing to do with obesity or body-fat. If we are to extrapolate these findings as a mechanism for high body-fat impairing hypertrophy, it seems like we’d have to assume that the differences between the groups were exclusively due to the divergence in training programs (rather than other factors such as sampling error, which can be notable in small-sample research with independent groups), the participants in the hardest training program were truly overreached (which seems unlikely, given that they experienced robust gains), the downsides of overreaching were largely or exclusively attributable to inflammation, the changes in local gene expression for cytokine receptors is an accurate indicator of actual systemic inflammation, and this same degree of inflammation (or a similar degree) would be induced by high body-fat alone. Menno’s article makes no attempt to tie any of those loose ends together into a cohesive or quantifiable argument.
Based on a fourth study, he suggests that “chronic inflammation, as measured by elevated C-reactive protein, also negatively correlated with muscle growth in a study of hospitalized elderly patients performing strength training.” That is true, but I can’t fathom generalizing those results to healthy lifters. This study wasn’t investigating people who volunteered to stay in a hospital for the duration of the study, or a study carried out using otherwise healthy folks who just happened to reside in a nursing home or assisted living facility. Rather, the subjects were elderly patients with a mean age of 84.8 years old, who were acutely admitted to the hospital for a variety of different medical conditions concerning enough to require multiple days of hospitalization. Notably, different medical conditions influence inflammation, physical function, and hypertrophy differently. Also notably, these patients were either seated or lying down for 21.9 hours per day, and inactivity is related to inflammation. Also very very notably, these participants completed an average of 4.9 training sessions, and the average time between pre-testing and post-testing was 6.2 days. Yes, we are looking at differences in muscle hypertrophy in totally untrained people over a span of roughly a week. I’m serious, follow the link.
As we’ll discuss later, Menno expressed skepticism about my article’s comparisons among American football players who play different positions, but this study is lumping together people who are acutely hospitalized for “pain or physical injury, respiratory disease, cardiovascular disease, genitourinary disease, endocrine disease, gastrointestinal disease, and other.” Menno’s interpretation seems to imply that the noteworthy characteristic distinguishing these folks from one another is inflammation levels, as his commentary fails to highlight the distinct medical conditions (and by extension, other health-related variables impacted by specific medical conditions) contributing to that inflammation. Once again, this article has absolutely nothing to do with obesity or body-fat, and the CRP levels observed are far higher than we could even dream of linking to a healthy person lifting weights at literally any body-fat level. For example, a 2015 study measured CRP levels in 674 obese participants (average BMI = 47.4) who were candidates for bariatric surgery, and found an average CRP level of 8.9mg/L. In this study on hospitalized patients, many had values well above 20mg/L, and some were up near 80mg/L. To help contextual those numbers, the normal reference range extends up to 10mg/L. Not that it’s worth putting too much stock in the results of this week-long study on hospitalized patients, but hypertrophy seemed to be fairly unaffected up to CRP levels of approximately 20mg/L – more than twice the average levels seen in obese bariatric surgery candidates. The figure below reflects not only the remarkably high CRP levels, but also the remarkably large changes in thigh lean mass occurring over about a week of training:
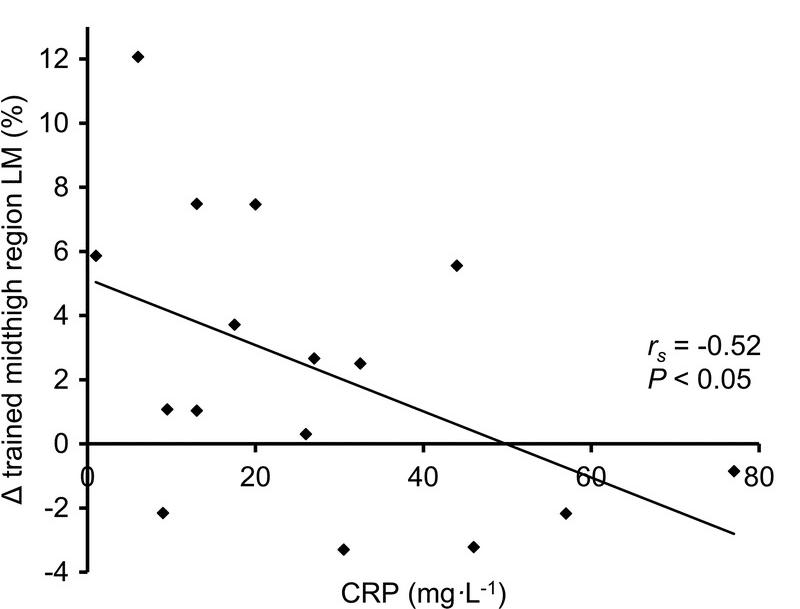
These four studies make up the bulk of empirical evidence presented in Menno’s inflammation article. It’s worth reinforcing that all studies included older participants (well into their 60s and beyond), and all samples included postmenopausal women; given the well-established link between aging and chronic inflammation, this should give us a little bit of hesitation when it comes to generalizing the findings to younger individuals. Menno’s inflammation article also cites a study by Mitchell et al to suggest that baseline inflammation is correlated with impaired gains in young, healthy, resistance-training subjects. In the discussion, the researchers reported: “We also found that pre-training resting IL-6 was inversely correlated with fibre hypertrophy.” What was that correlation? I have no idea. It isn’t reported anywhere in the results. Were other baseline measures of inflammation, such as TNF-alpha and CRP, correlated with hypertrophy? No idea. They were measured, but the study reported no other relationships between any of the baseline blood measurements and hypertrophy. I don’t like when findings appear out of nowhere in the discussion, especially when the methods section doesn’t say that the relationship is going to be analyzed, and the relationship is not reported in the results, and other similar analyses are inexplicably absent from the paper. Given that this analysis isn’t reported in the methods and isn’t reported in the results, it’s impossible to know the magnitude of this relationship, and hard to know if it was a planned analysis, or a fairly random correlation that popped up from poking around the data in an exploratory manner. If the latter, it may well be a false positive. If this was a planned analysis, one would assume it would also be carried out for TNF-alpha and CRP, and one would assume that those relationships would’ve been reported if they were also significant. As a result, this study certainly fails to present a rock-solid case that baseline inflammation was an important factor moderating hypertrophy (it appears that only one of three inflammatory biomarkers was associated with hypertrophy, and we don’t have quantitative information about the association), and also fails to connect inflammation to body-fat in any way.
Furthermore, this study fails to make a case for elevated systemic inflammation affecting hypertrophy, even if we take the correlation between baseline IL-6 and hypertrophy at face value. Normal IL-6 levels are <5pg/mL. The baseline IL-6 levels in the Mitchell study were 2.2 ± 0.6pg/mL. So, tacking 2 standard deviations onto the mean, most of the subjects likely had IL-6 levels between 1pg/mL and 3.4pg/mL – comfortably within the normal range. In other words, if we were to accept that baseline inflammation levels, as indicated by IL-6, influence your hypertrophic response, we’d also need to accept that they can have a notable negative impact on hypertrophy before a lab test would even begin to suggest that your inflammation levels were outside the normal range. If that’s the case, this implies a pretty strong effect that should be easily detectable in other studies; on the contrary, the other studies reviewed here have failed to detect this effect.
I’d like to highlight a few overarching themes in this inflammation line of argument. First, the supporting data is almost entirely separate from body-fat, and much of the evidence comes in the context of circumstances with high inflammation levels that are independent of body-fat (acute hospitalization, extreme inactivity, untrained status, and purported overreaching). Second, the supporting data is inconsistent in the studies reporting multiple markers of inflammation. Some evidence pins the blame on TNF-alpha while CRP and IL-6 are unimpactful, whereas other evidence pins the blame on IL-6 while TNF-alpha and CRP are unimpactful. One study found significant overreaching-induced changes (again, absolutely nothing to do with body-fat) in expression of only two of the five inflammation-related genes tested, and the inflammation only seemed to impact lower-body outcomes. One study suggested that baseline IL-6 was the big factor to worry about, while another found that baseline IL-6 was not negatively correlated with subsequent gains, and another found that baseline IL-6 did not differ between the responders and non-responders, while suggesting that non-response was attributable to excess inflammation. If you’re going to claim that a mechanistic explanation is so air-tight that it can be safely generalized from totally different contexts (for example, from sedentary people with serious diseases to active people with higher body-fat levels), it needs to be more consistent than that, and you have to acknowledge that the dose makes the poison. This argument could be strong if it suggested that a given amount of systemic inflammation was reliably associated with hypertrophy impairment, and that a given level of body-fat reliably caused a similar magnitude of systemic inflammation in otherwise healthy people who regularly lift weights. So far, that evidence hasn’t been presented. To my knowledge, such literature does not yet exist, which is why I initially set out to find evidence of young, healthy, active people struggling to gain lean mass because their body-fat was too high. After all, if we aren’t even observing the effect, we don’t need to drive ourselves crazy looking for a cause. As outlined in my article, I’m not seeing enough evidence of this scenario playing out to necessitate the identification of a cause.
I want to be very clear about something: I have no doubt that excess inflammation beyond a certain threshold is bad news for hypertrophy. However, I am very confident that there’s a wide range of “normal-ish” inflammation levels, and that fluctuation within this range is unlikely to modify hypertrophy to a noticeable magnitude. I don’t think it’s a coincidence that most of the human evidence Menno provided that was related to inflammation blocking hypertrophy was from studies in older adults with relatively low training status and activity levels at baseline, as inflammation is independently impacted by age and activity level. While Menno does provide evidence that inflammation has been unfavorably associated with body composition changes in some contexts, there are a few big hurdles to clear before this can be designated as an explanatory mechanism by which body-fat impairs hypertrophy, especially in active populations. In order to do so, you’d have to show that high body-fat levels independently and reliably increase inflammation enough to meaningfully impact hypertrophy in young, otherwise healthy people, and that this inflammation isn’t insufficiently attenuated when that individual begins to engage in resistance exercise, which reduces chronic inflammation levels independent of weight loss.
Menno’s rebuttal doesn’t come anywhere close to doing that. Until those loose ends get tied up, this argument is about as evidence-based as claiming that eating a handful of almonds is lethal. Cyanide is lethal, and almonds contain cyanogenic glycosides that release hydrogen cyanide when hydrolyzed. When framed that way, it seems clear that I should probably present empirical data indicating that almond consumption is actually dangerous, or at least mention the lethal dose of cyanide, the type of almond we’re talking about, and how much cyanide we’re getting from that type of almond, instead of assuming that A leads to B and B leads to C, with no regard for magnitudes, context, or empirical data connecting the purported cause to an observed effect. Just to be clear, sweet almond cultivars are totally fine, and as discussed in the original p-ratio article, people with high body-fat are making plenty of gains. The almond example is an exaggeration, and Menno’s hypothesis of course has more plausibility than this example, but until the hypothesis is accompanied by some quantified metrics that pertain to the context it’s being applied to (i.e., how much body-fat, how much inflammation, and so on), it’s more of a vague notion than a theoretical model.
Topic: Protein Balance
In this section, Menno presents differing interpretations of the literature comparing acute muscle protein synthesis responses in lean and obese participants. By now, it should be clear that I’m not super enthusiastic about making inferences about hypertrophy in lifters based on data that involves no resistance training stimulus whatsoever. So, this really comes down to two studies.
Menno first states, “Beals et al. (2018) found reduced myofibrillar protein synthesis levels in overweight vs. lean individuals after a strength training workout combined with protein consumption. There was no anabolic resistance to sarcoplasmic protein synthesis.” We’ll take a look at the actual data, but here’s a quick primer on the study design: there was a lean group and an obese group. Sarcoplasmic and myofibrillar protein synthesis rates were measured in “basal” conditions (relaxing and fasted), and after consuming lean ground pork (~36g protein). Before consuming the pork, they completed resistance training with only one of their legs. So, measurements were taken on both their untrained leg (to make inferences about protein feeding only) and their trained leg (to make inferences about the combination of protein feeding and resistance exercise). The results are presented in this figure:
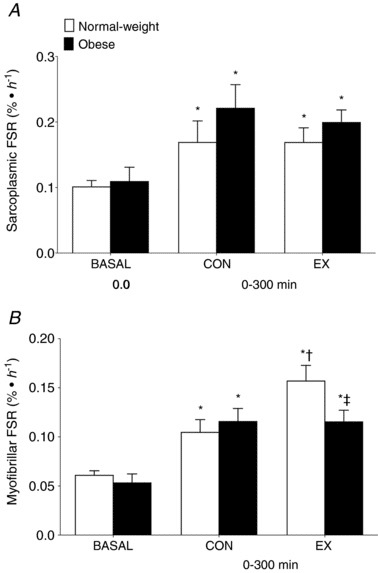
First, let’s look at the control leg. Both the obese and lean subjects had significant increases in both saroplasmic (top figure) and myofibrillar (bottom figure) protein synthesis rates. Now we’ll look at the additive effect of resistance exercise. When we compare the lean control leg to the lean exercise leg, we see that sarcoplasmic protein synthesis was totally unaffected by resistance training, but myofibrillar protein synthesis increased significantly. When we compare the obese control leg to the obese exercise leg, we see that sarcoplasmic protein synthesis was actually a little lower after exercise, and myofibrillar protein synthesis was totally unaffected. If we are to take these values literally, and at face value, and assume that they provide us with meaningful information about subsequent hypertrophy, it would seem that obese people who lift would achieve no greater hypertrophy than obese people who do not lift, assuming that protein is matched. I reject that idea unequivocally based on volumes and volumes of longitudinal studies, which makes you wonder exactly how readily we should assume that these findings give us firm conclusions about hypertrophy potential.
The Beals study also provides some uncomfortable contradictions within Mennos’ argument. As the authors note in the discussion, “What is noteworthy is that this blunted post‐exercise myofibrillar protein synthetic response with obesity occurred despite a lack of clear differences in habitual physical activity levels or systemic inflammation between the groups.” If Menno’s argument is to remain intact and non-contradictory, I would speculate that he would just as readily advocate for some serious caution when interpreting these data, because if taken literally, they take his primary proposed mechanism (inflammation negatively influencing hypertrophy) off the table.
The second study was by Hulston et al, which showed that rates of mixed muscle protein synthesis were similar among obese and leaner individuals in response to resistance exercise. Menno suggests that this study appears to contradict the findings of Beals et al due to a methodological difference. Menno implies that obesity impairs the myofibrillar protein synthesis response, but not the sarcoplasmic protein synthesis response. As a result, the idea is that Hulston “missed” the important stuff by looking at mixed muscle protein synthesis instead of looking at sarcoplasmic and myofibrillar synthesis rates separately. If we look back at those figures from the Beals study, this interpretation does not fit the data. Hulston was specifically looking at the response to resistance exercise, and resistance exercise did not stimulate an increase in myofibrillar or sarcoplasmic protein synthesis in the obese subjects in the Beals study. In fact, sarcoplasmic protein synthesis was slightly lower in the trained leg than the untrained leg in obese participants. Perhaps a more suitable explanation for the different findings between these studies relates to participant characteristics. While Beals and colleagues sampled participants who “were considered as insufficiently active based on a Godin leisure‐time exercise questionnaire,” Hulston et al state: “Participants in both groups reported taking part in at least 3 × 30 min of moderate‐intensity physical activity per week. Young, physically active participants were recruited for this study as both aging and inactivity are known to contribute toward anabolic resistance and muscle atrophy. Hence, this study focused on obesity, separate to these other known risk factors.” In other words, the differences between these studies could be explained by the fact that Hulston purposefully recruited active subjects, while Beals recruited sedentary subjects, suggesting that the findings of the Hulston study may be more generalizable to lifters. When taken literally and assumed to represent hypertrophy potential (more on this soon), the Beals results don’t make a ton of sense, because they imply that obese individuals are effectively immune to hypertrophy induced by resistance exercise. They also imply that systemic inflammation has little to do with the attenuation in protein synthesis.
In his rebuttal, Menno states: “Trexler & Nuckols largely write off these 2 study findings [Beals et al and Hulston et al] as ‘inconsistent’, but I’d say the overall trend in the literature is decidedly negative for overweight individuals.” In fact, the inconsistencies in this literature are quite clear. There is a review paper by Beals et al that presents plenty of evidence that obese individuals have a smaller muscle protein response to protein feeding than leaner individuals. I wouldn’t get super worked up about this literature; we’ve already covered all of the studies that involved a resistance training component, and all of the others in this review involved untrained participants in a rested state. Anyway, the review by Beals et al indicates that obese people have a blunted muscle protein synthesis response to dietary protein. However, the intervention by Beals et al indicates that obese people actually had equal or slightly greater muscle protein synthesis than leaner people in response to protein alone, whether you’re looking at myofibrillar or sarcoplasmic protein synthesis. However, they did find that obese people had a completely blunted muscle protein synthesis response to resistance exercise (literally no increase whatsoever for myofibrillar or sarcoplasmic synthesis), so they pinned the blame on an impaired response to resistance exercise. However, Hulston et al found that obese people not only had a robust protein synthesis response to resistance exercise, but the response was not significantly different than the response observed in leaner individuals. So, we’ve got three main pieces of literature in the discussion, and each one directly contradicts another.
If you had one study saying that a supplement caused liver damage with no adverse effect on the kidneys, and one study saying that a supplement caused kidney damage with no adverse effect on the liver, you would not have unanimous agreement that the supplement is bad. You’d have an inconclusive literature with two findings that are directly contradictory.
When it comes to the protein synthesis studies involving resistance exercise, the intervention by Beals et al is the only one (out of two) that generally supports the p-ratio idea. However, if we are suggesting that this finding has any relevance to the topic at hand, we must necessarily be implying that the acute muscle protein synthesis response is indicative of hypertrophic potential, and can be extrapolated accordingly. If we reject that implication, then the study is of no consequence whatsoever for the current discussion. If we accept that implication, the findings of the study have no face validity whatsoever, as the resulting implication would be that obese people who lift will experience slightly less, or maybe equivalent, amounts of hypertrophy when compared to obese people who don’t lift.
As I mentioned previously, there is a review paper by Beals et al that presents plenty of evidence that untrained obese individuals, in a rested state, have a smaller muscle protein response to protein feeding than leaner individuals. When I acknowledged this literature in my original p-ratio article, I mentioned that a study by Murton et al suggested that the blunted protein synthesis response to ingested protein (again, in the absence of resistance training) might be counteracted by a reduction in muscle protein breakdown. Menno had a different interpretation of the paper; I don’t agree with his interpretation, but it is a fair and defensible interpretation. The nature of our differing perspectives basically comes down to the net balance of two figures; one shows the protein synthesis response to protein ingestion, the other shows the protein breakdown response:
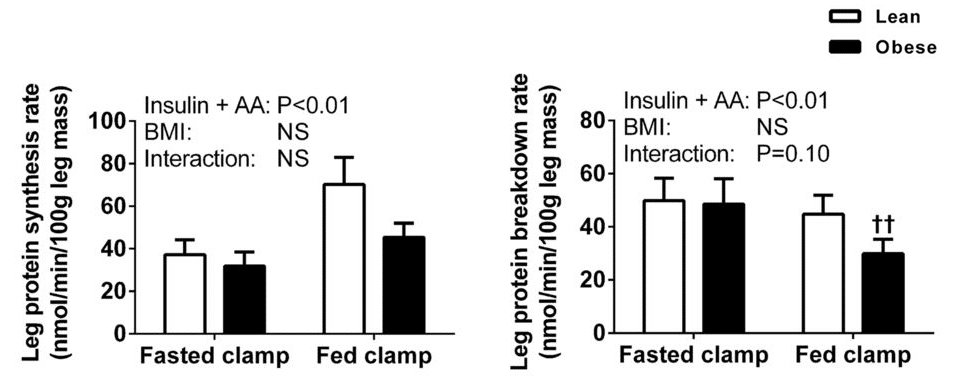
As we can see, when participants consumed protein (that is, went from fasted to fed) protein synthesis increased more in lean participants than in obese participants. At the same time, protein breakdown decreased more in obese participants than in lean participants. As the researchers put it, “Our results suggest that the rate of LPB in the postprandial state was lower in obese than in healthy-weight volunteers. Therefore, the inability of amino acids to stimulate MPS in the obese appeared to be offset largely by a concomitant decline in the rate of LPB, culminating in the magnitude of the change in net phenylalanine balance between the postabsorptive and postprandial states being equivalent among both healthy-weight and obese subjects.”
To be fair, they probably should have said “not significantly different” rather than “equivalent,” given that equivalence testing did not occur. In addition, the between-group difference in protein synthesis was largely offset by the between-group difference in protein breakdown, but not entirely. The lean group was in slightly more positive protein balance, although this difference between groups was not statistically significant:
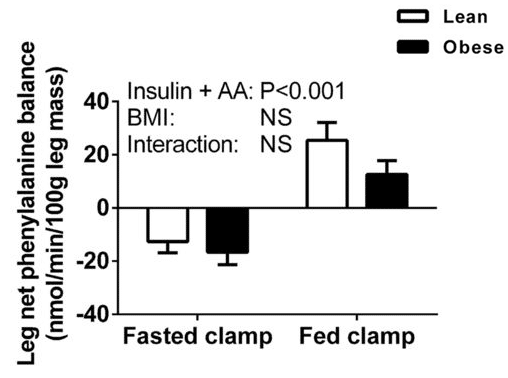
So, the interpretation of these results comes down to a judgment call about whether or not you consider this non-significant difference to be physiologically meaningful. I don’t, and Menno does, but that’s an entirely fair interpretation.
Before moving on, I should also note that this study actually “stacked the deck” a bit (which is totally fine) in favor of the inflammation hypothesis. Participants were older than the Beals and Hulston studies that included a resistance training stimulus, and when they recruited the groups for this study, obese participants were required to have elevated inflammation (CRP > 1.35 µg/mL), and lean participants were required to have low inflammation (CRP < 1.35 µg/mL). If you were lean and had high inflammation, or if you were obese and had low inflammation, you were prohibited from participating. So, if a study was going to show noteworthy effects of obesity-related inflammation, it would be a study like this, which sampled older and relatively inactive individuals and artificially ensured that groups would differ on the basis of inflammation levels. The fact that the results are open to interpretation is, in a way, pretty underwhelming support for the inflammation-focused hypothesis about hypertrophy and p-ratios.
However, even if I were to concede that Menno’s interpretation of the Murton study was unequivocally correct, and that these findings were physiologically meaningful, that would lead us to two big problems. The first problem is that we’ve still got only two studies comparing acute rates of post-exercise muscle protein synthesis over a timespan of a few hours, and both studies were conducted in untrained subjects completing a single resistance training session. I don’t like pasting huge blocks of texts, but Dr. Jorn Trommelen summarized this so perfectly that I want to leave his words from a recent review paper intact:
“While no correlations were observed [in this study] between post-exercise myofibrillar protein synthesis rates following the initial exercise bout and muscle hypertrophy following prolonged exercise training, strong positive correlations were observed between myofibrillar protein synthesis rates assessed over a 48-h period at 3 weeks and at 10 weeks of training and the increase in muscle mass. The initial exercise bout resulted in considerable muscle damage, but exercise-induced muscle damage was attenuated at 3 weeks of training and almost completely absent at 10 weeks. Therefore, it appears that the myofibrillar protein synthetic response to a single bout of unaccustomed exercise may be at least partly a response to muscle damage and directed at tissue repair rather than muscle hypertrophy. After the first couple of days or weeks of training, post-exercise myofibrillar protein synthesis rates may be more reflective of the net changes in muscle mass, i.e., muscle hypertrophy.”
Jorn’s main points were that muscle protein assessments may be reasonably predictive of hypertrophy, but only when the actual assessment period is pretty long (48-72 hours rather than a few hours) and subjects are reasonably accustomed to the training stimulus. So, while the studies on acute muscle protein synthesis that lacked a resistance training component aren’t particularly relevant for our inferences in lifters, the two studies that included a resistance training component also have generalizability issues because they put untrained subjects through novel resistance training protocols and protein synthesis responses were only observed for a few hours post-exercise. So, as I noted in the original p-ratio article: “even if this mechanistic relationship was conclusively identified in these acute scenarios, it would take another leap of faith to extrapolate that to long-term body composition changes and hypertrophy over the course of a longitudinal resistance training program.”
The second problem is that this creates a very uncomfortable discrepancy within Menno’s own theoretical model. As Menno states himself, “our maximum muscular potential is higher at higher body-fat levels. Our bodies seem to allow us to carry more muscle when we also carry some more fat.” I agree with this statement, but it insists that we accept a curious contradiction: higher body-fat pushes us into a state of less positive protein balance, which can be extrapolated to make inferences about impaired potential for hypertrophy, but higher body-fat is also a necessary prerequisite for achieving our maximal degree of muscularity. Hypertrophy is simply a manifestation of positive protein balance over time, and achieving our maximum degree of muscularity is simply a manifestation of consistently achieving hypertrophy over time. Or, presented visually:
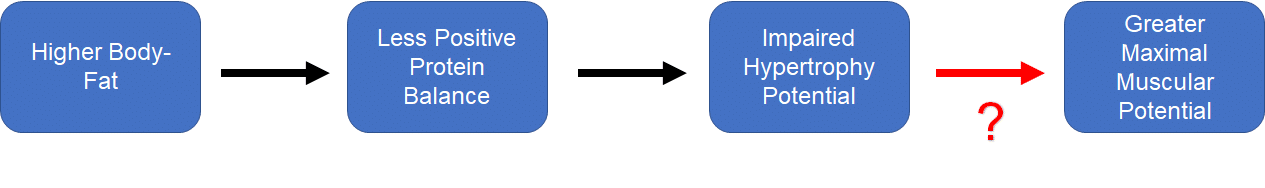
How might we explain the red question mark? That is, how might high body-fat independently impede hypertrophy while being a prerequisite for achieving maximal hypertrophy over time? Based on the p-ratio hypothesis, one might suggest that body-fat does impair hypertrophy, and that this impairment must be overcome by an even larger caloric surplus, which would require that people must gain more body-fat for a given magnitude of lean mass gain. I’ve never seen any data to support this contention, and I shared multiple pieces of contradictory evidence in my original article.
Finally, Menno reinforces his argument by citing a study about acute protein oxidation, but I think that particular evidence is weak enough that it really doesn’t require a thorough rebuttal on my end. He cites a study showing that after acute consumption of a meal, obese people oxidized more protein than leaner people. It’s noteworthy that the obese participants received a meal with more total protein, and no resistance training was involved. Since we don’t store protein the same way we store carbohydrate or fat, I feel like it’s pretty intuitive that they received more protein than could immediately be utilized for protein synthesis, so they oxidized the rest. A rebuttal to that point might be an argument suggesting that the obese subjects should have received more protein, as they had more fat-free mass. However, even if both groups received the same exact amount of protein, I’m not convinced that an acute whole-body protein oxidation measurement can tell us much about the topic at hand. The implication is that acute nitrogen excretion and energy expenditure can be used to predict protein oxidation, and protein oxidation serves as a proxy for acute whole-body protein balance, which serves as a proxy for acute muscle protein balance, which we can use to make inferences about long-term maintenance of muscle mass.
You’re going to have a tough time convincing me that acute post-meal protein oxidation, estimated via urinary nitrogen excretion and indirect calorimetry, can be used to make inferences related to long-term hypertrophy potential. Even if post-meal protein oxidation could be used as a perfect proxy for acute whole-body protein synthesis, muscle mass contributes only 25-30% to whole-body protein turnover, and even if acute whole-body protein turnover were a perfect proxy for acute muscle protein synthesis, acute muscle protein synthesis correlates quite poorly with hypertrophy. I think the authors of a recent paper state things quite effectively: “…more amino acids are directed towards oxidation after ingestion of larger amounts of protein. It should be noted, however, that the postprandial rise in whole-body protein synthesis rate, greater whole-body protein balance, and whole-body amino acid oxidation do not necessarily reflect the anabolic response in skeletal muscle tissue.”
Topic: Recovery Capacity
This is a curious section of the rebuttal; it cites several studies ranging from mostly irrelevant to totally irrelevant, in areas of the literature where relevant studies exist. First, Menno states “In animal and in vitro research, we see significant detrimental effects of obesity on muscle recovery capacity.” The review he cites focuses largely on research utilizing rodents possessing notable gene mutations or consuming high-fat chow. As noted in the previously reviewed study by De Sousa et al, it’s possible that high-fat chow has hypertrophy-impairing effects that are independent of the obesity it induces in rodents. As for the mutant rodent lines, they generally lack a leptin receptor or lack the ability to produce leptin. There is evidence that leptin itself may have anabolic effects, and that leptin infusion directly impacts anabolic and catabolic processes in the muscle tissue of leptin-deficient mice. Nonetheless, the broader caveat is that changing a hugely important gene rarely has exactly one effect on physiology, and often has “ripple effects” that influence a wide range of physiological processes in the body. So, you have to put some effort into justifying the idea that findings in genetically altered mice actually generalize to wild mice, let alone healthy, resistance-trained humans with normal leptin signaling. If this rodent data were all we had available, then we’d have to make do with it, but that’s simply not the case.
In his rebuttal, Menno states that “A human study on handgrip strength testing found evidence of reduced stress tolerance and greater fatigability in obese vs. lean individuals.” In this study, the handgrip measures of strength endurance obtained during 20+ minute continuous bouts with 30% of maximal voluntary contraction force (which have dubious generalizability to the typical lifting program) were not significantly different when comparing obese and non-obese participants in a control condition (when both groups were just exercising). The results simply indicate that the obese individuals had a tougher time during the “stress” condition, which involved doing mental math problems while lifting. So as long as you’re periodizing your math problems effectively (I recommend an undulating system, in which math problems vary within microcycles and occur outside of resistance training bouts), you’re good.
Menno also states that “poor carb tolerance has also been directly linked to poorer adaptations to training, at least during endurance exercise.” The study cited by Menno evaluated endurance training adaptations, which are entirely distinct physiological processes from muscle hypertrophy. More importantly, this study definitely doesn’t support his claim. None of the groups were carb-intolerant, and their baseline metrics related to insulin sensitivity and carbohydrate tolerance were virtually identical. The first sentence of the discussion reads, “In this study, we have evaluated the response to an exercise intervention in healthy, glucose-tolerant FH− and FH+ subjects with the two groups similar in regard to age, BMI, and V̇o2 peak.” The paper also states “The FH+ and FH− groups were similar in age, BMI, V̇o2 peak, habitual activity, and glucose tolerance and insulin sensitivity (all with normal fasting glucose, OGTT, and homeostatic model assessment-insulin resistance).” Menno’s claim about this study is not related to anything the study actually investigated.
As I’ve alluded to already, this section of the rebuttal could have been strengthened by focusing on some much more relevant research. One study compared obese and lean individuals completing reps to failure on multiple sets of chest press, leg press, lat pulldown, leg extension, overhead press, hamstring curls, biceps curls, calf raises, and triceps extensions. While the raw repetition values are curiously high for all groups and conditions, the data certainly don’t point toward greater fatigability in obese individuals when compared to leaner individuals. Similarly, a 2013 review assessing four studies utilizing isokinetic dynamometry concluded that there was insufficient evidence to assert that muscular fatigability is increased in obese individuals.
The section ends with the following figure, which documents the “plausible mechanisms by which being overweight may negatively affect muscle recovery.”
Plausible mechanisms brought us the hits such as “carbs cause obesity,” “protein damages kidneys,” “protein causes bone loss,” “foods with high glycemic index values give you diabetes,” “dietary cholesterol gives you heart disease,” “vegetables are poison because of anti-nutrients,” “creatine causes virtually all bad things,” and so on. That is, many plausible mechanisms sound good until empirical data render them less plausible, so dropping a plausible mechanism and walking away without providing some degree of empirical support comes up short when trying to extrapolate mechanistic data to a specific context (unless that extrapolation is labeled as purely speculative and uncertain, and the empirical support is absent because no directly or indirectly relevant empirical support exists).
If Menno wanted to make a stronger case in this section, he could have cited any of the studies that actually directly demonstrate the effects of higher body-fat levels on recovery following resistance exercise. Greg and I were able to find four studies pretty easily that compare recovery after resistance exercise in lean (or normal BMI) subjects versus subjects with higher body-fat levels (or higher BMIs; 1, 2, 3, 4). All four demonstrate faster recover times in the lean/normal BMI subjects. However, even after strengthening Menno’s argument for him, it’s still not a particularly strong argument. Again, we’re interested in the effects of higher body-fat levels in lifters. The subjects in these four studies were untrained, and it shows. For example, in both the lean and obese subjects in the first two studies, isometric strength was still suppressed below baseline values four days post-eccentric exercise, and creatine kinase levels were still climbing at four days post-training. However, we also know that due to the repeated bout effect, muscle damage responses following resistance training decrease and recovery rates increase after people accumulate a bit of training experience. In other words, there is actually good data suggesting that higher levels of body-fat interfere with recovery from resistance exercise in untrained individuals, but that still doesn’t tell us much about the effects of higher body-fat levels on muscle recovery in lifters.
Topic: Anabolic Hormones
In the rebuttal, Menno discusses body-fat percentage and fertility. As he notes in his article, this is not relevant to the argument put forward in my article. However, I was taken aback by his assertion that exactly 10.7% body-fat is the ideal body composition for fertility in males, and 21.5% body-fat is the ideal body composition for fertility in females, so I feel an obligation to comment on that, just to make sure readers don’t take that number seriously and use it to guide important family planning decisions. Menno derived that number from this study, based on his interpretation that “subfecundity” (a waiting time of more than 12 months [or 6 months] to achieve a pregnancy that resulted in a live birth) was lowest in people with BMIs <18.5, and increasingly worse in people with BMIs from 18.5-24.9, 25-29.9, and >30 (notably, the study didn’t actually report body-fat percentages. More on that later). The table presenting these results is included below; note that we are most interested in the marginal values for men (inside the red box) and women (inside the green box).
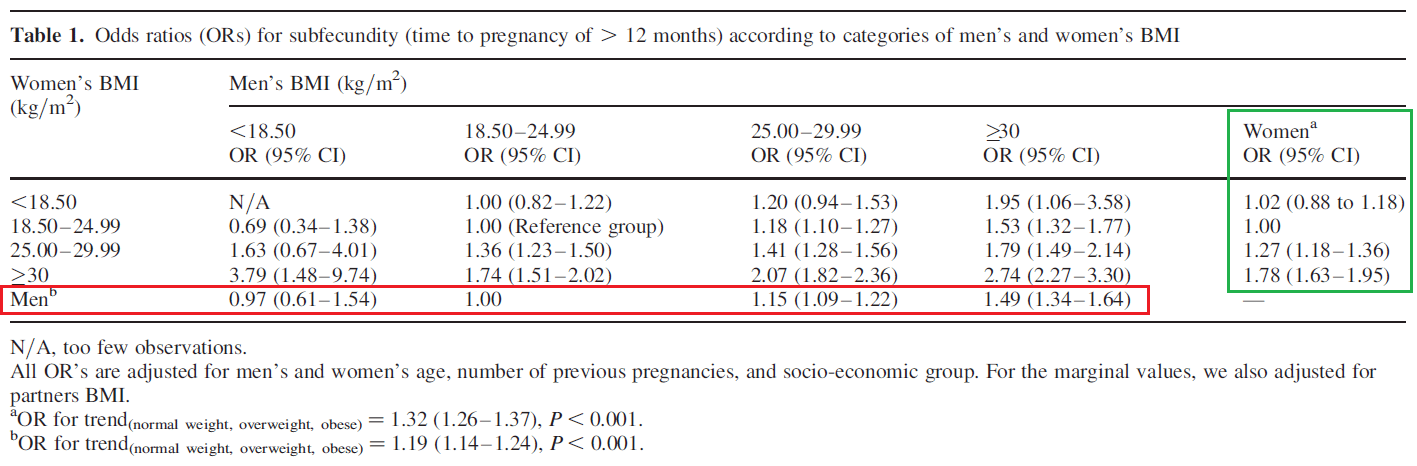
If you look at the data for men in this study, their odds ratio for subfecundity at BMI < 18.5 was 0.97, with a 95% confidence interval spanning from 0.61 to 1.54. For reference, if the value was 1.0, it would be identical to the group with BMIs between 18.5-24.9, and if the confidence interval contains 1.0, the groups cannot be said to have significantly different odds ratios. The p-value corresponding to this odds ratio was not reported, but it can be calculated, and it’s greater than 0.9 (that is, more than 18-fold higher than the p-value you’d use to suggest that odds ratios become different when you drop from one group [BMI 18.5-24.9] to the next [BMI < 18.5]). In other words, male subfecundity was practically identical for these BMI categories when controlling for all the important confounders in the model.
You could argue that you don’t care about statistical significance, and this 0.03-unit odds ratio increase still matters to you. However, you’d immediately contradict yourself with the female data in the same study. The subfecundity odds ratio value for women with BMI < 18.5 was actually 0.02 units higher than women with BMI 18.5-24.9 (1.02 vs. 1.00). But again, this difference was not even close to statistically significant, with a 95% confidence interval spanning from 0.88 to 1.18. So, a more defensible interpretation of this study would be that subfecundity starts to increase when BMI begins to exceed 25. But that brings us to a good question: how does this study yield an estimate of 10.7% body-fat for males? That is, how does “any BMI under 18.5” become “exactly 10.7% body-fat?”
Greg reached out to Menno, and Menno was kind enough to explain, which we both appreciate. The calculation involved assuming an age of 21 years and using the formula from this study to convert BMI to body-fat percentage. Using the formula in that study, a BMI of exactly 18.4 translated to a body-fat percentage of 10.7% in males and 21.5% in females. If we accept that subfecundity becomes meaningfully impaired above a BMI of 18.4 in males, then we should say that optimal male body-fat for fertility is ≤10.7% rather than exactly 10.7%, because BMI was lumped into categories in the study itself. However, as noted previously, subfecundity didn’t start to become elevated (in terms of clinical relevance or statistical significance) until BMI got above 25 in males (again, in females, subfecundity was nominally reduced when comparing BMIs of <18.5 to BMIs of 18.5-25, so there doesn’t seem to be any reasonable way to justify the 21.5% body-fat recommendation in females, even if you do allow for incredibly liberal statistical inferences). So, if you’re comfortable assuming that we can convert BMI to a body-fat percentage with reasonable accuracy, then the data from that study would indicate that subfecundity starts to get meaningfully impacted as body-fat percentages start getting above 18.5% for males and 29.3% for females. Again, that has nothing to do with what we’re talking about, but I think the correction was warranted since those numbers entered the discussion.
After that, Menno says “Some people argue differences in testosterone level do not influence muscle growth, but I disagree with that.” Notably, the paragraph contains no references. An assertion made without evidence can be rejected without evidence, but I would still encourage readers to check out a fantastic article by James Kreiger about how testosterone fluctuations within the physiological range influence hypertrophy. In it, James provides a compelling and heavily referenced argument that fluctuations within the normal range have pretty modest impacts on hypertrophy and fat-free mass. Menno rightfully points out that many such arguments draw upon comparisons between individuals rather than within individuals, which is a limitation. But what Menno fails to provide is evidence that lifters can increase their testosterone by a magnitude that is theoretically sufficient to meaningfully affect hypertrophy by losing fat (or, better yet, empirical evidence of this fat loss-induced, testosterone-mediated hypertrophy actually occurring).
On the contrary, Eriksson and colleagues completed a Mendelian Randomization Analysis of five large studies reporting BMI and testosterone levels. The difference in testosterone concentrations between the “overweight” (BMI of 25) and “obese” (BMI of 30) cutoffs seems to be approximately 13%. In other words, if you’d have a testosterone concentration of 600ng/dL at a BMI of 25, you could expect to have a testosterone concentration of 522ng/dL at a BMI of 30. It is true that you can find research showing that very large differences in testosterone levels can impact hypertrophy. For example, Bhasin’s famous testosterone study found a clear dose response relationship between exogenous testosterone administration and hypertrophy, and Kvorning et al found that when you suppress testosterone levels from the middle of the normal range (~650ng/dL) to basically nothing (~30ng/dL) with testosterone-suppressing drugs, gains in leg lean mass following resistance training may be a bit smaller (0.37kg increase vs. 0.57kg increase in subjects without suppressed testosterone levels). However, you’d be hard pressed to find a study where a mere 13% difference in testosterone makes a notable difference in hypertrophy. Of note, 5 BMI points suggest a difference in body-fat percentage of approximately 8.5%, so if you decide to cut a bulk off at, say, 23.5% instead of 15% body-fat, or even 28.5% instead of 20% body-fat, you probably won’t be dealing with a large enough change in testosterone levels for the difference to be physiologically relevant. It’s very possible that obesity-induced suppression of testosterone could become an issue at sufficiently high body-fat levels, but our assumption when we write an article for lifters is that most people don’t intend to extend a bulk until they hit, say, 40% body-fat.
Topic: Indirect Lines of Research
This is basically a “rapid-fire” section in which Menno presents a few criticisms on a variety of different topics, so I’ll respond to each as concisely as possible.
Menno states: “Trexler & Nuckols draw on a commendable body of indirect research to investigate if high body-fat levels interfere with our ability to make lean gains. However, I think this literature is simply too indirect and confounded to draw any conclusions from.”
I disagree with this contention. While I acknowledged the limitations of each body of literature, it seems hard to imagine that despite the innumerable resistance training studies that have been published to date, sampling thousands of lifters with a wide range of body-fat levels, we simply cannot obtain a glimpse into whether or not body-fat percentage is an influential predictor of body composition changes. Moreover, I contend that data on football players is more relevant to lifters than data on completely sedentary, hospitalized octogenarians.
Menno states: “The finding that the many of the most muscular athletes are the fattest ones says nothing about nutrient partitioning. It simply reinforces the quite well-established finding that our maximum muscular potential is higher at higher body-fat levels. Our bodies seem to allow us to carry more muscle when we also carry some more fat.”
In a vacuum, this is true. You could assume that most of these athletes started very lean, and their p-ratio got terrible as they gained weight. Conversely, you could assume that most linemen and super heavyweights and other big strength athletes tended to carry a decent amount of extra body-fat before they got really serious about muscle growth (this has been my experience, but it’s still an assumption). Nonetheless, as I mentioned in the original article, “impaired p-ratio during bulking” is essentially synonymous with “impaired hypertrophy.” If you overfeed, weight gain will occur, and the p-ratio will simply vary based on how much lean mass you were able to add. So, an impaired p-ratio should indicate that lifters with higher body-fat percentages should either struggle to add lean mass in general, or at minimum should require a disproportionately large increase in fat mass per unit of lean mass gained. When we look at sumo wrestlers climbing the ranks in Hattori et al, this pattern is not observed. When we look at body composition changes in college American football players across their first year at the college level (1, 2) or a full four-year career (3), an opposite pattern is observed. When we look at individual-level data from seven different resistance training studies from a number of different laboratories, an opposite pattern is observed: the athletes with the most body-fat either gained the same amount of muscle as the leaner athletes or more muscle than the leaner athletes, and lost the same amount of fat or more fat than the leaner athletes. The moment the evidence in favor of the hypothesis (worse p-ratios with higher body-fat levels) overshadows the evidence contradicting the hypothesis, I will jump on board with the idea, but I haven’t seen any evidence supporting it. If the ultimate outcome of me writing my p-ratio article is that people finally dig up some evidence to actually support this widely accepted hypothesis, and I end up being totally wrong, then that’s still a big win in my book. In the meantime, we return to the question that was posed earlier: how do we navigate Menno’s hypothesis indicating that higher body-fat pushes us into a state of negative protein balance, which is extrapolated to make inferences about impaired potential for hypertrophy, while acknowledging that higher body-fat is also a necessary prerequisite for achieving our maximal degree of muscularity?
Menno states: “Comparisons between different positions of American football players are confounded by numerous factors. First, there’s huge selection bias here. For example, the prototypical hardgainer probably doesn’t want to be or is going to be selected to be a lineman.”
This is true, which is why we sought out individual-level data from seven separate studies sampling untrained and resistance trained members of the general population, and the results from our analysis line up quite well with the data from football players.
Related to the football data, Menno states: “Few of these studies have any diet controls and the rare ones that do typically suffer from atrocious adherence rates.”
Menno cited very few longitudinal studies in lifting humans, but I checked the five studies he cited to establish the link between inflammation and hypertrophy. One study asked participants to jot down their diet from time to time, but zero of them implemented any type of diet control, and one study had an attrition rate of 43.6%. In addition, studies on college sports teams tend to have great adherence; teams generally have constant events (study groups, meetings, practices, workouts, and so on), and the lab visit just becomes one more event that your entire peer group is doing. In well-funded college football programs, a lot of teammates eat together at the same times, in the same facility, with the same meal options, which are often curated by the same dietetics staff. It’s not a substitute for strict dietary control by any means, but in a resistance training literature in which dietary control is an extreme rarity, I’d venture to say that college football studies are as good as (or better than) a typical training study when it comes to minimizing major nutritional confounders.
Menno states: “Post-competition weight regain is again majorly confounded by several factors. First and foremost, after a competition many competitors binge the crap out of themselves. They’re not exactly in the ‘lean bulking’ mindset. Plus, this population actually likely has too low a body-fat percentage: their hormonal health is shot.”
I agree that post-competition p-ratios are actually threatened, not enhanced, by low body-fat. More importantly, this preference for fat regain after weight loss isn’t restricted to physique athletes at shredded body-fat levels; there’s plenty of evidence showing that weight regain following weight loss is characterized by a p-ratio that is either worse or the same as before weight loss occurred. This is a pretty huge deal; everyone talks about their p-ratio as if they can longitudinally modify it (for example, one might cut to 11% body-fat in an effort to potentiate their future gains). If weight loss is most likely to have a neutral or negative impact on your p-ratio when you transition from a cut to your next bulk, then the utility of this hypothesized p-ratio concept is severely diminished. The underwhelming results from weight regain studies across a wide range of body-fat levels, combined with the evidence pointing toward one’s p-ratio being a largely intrinsic characteristic that varies from person to person, shifts this p-ratio conversation from “you should cut to improve your p-ratio” or “you should minimize fat gain to avoid messing up your p-ratio” to “you should hope you were born with a terrific intrinsic p-ratio.” I agree with the third statement, but we have no empirical evidence for the first two (in people who are lifting weights).
Topic: Meta-Analysis of Longitudinal Resistance Training Studies
In my original p-ratio article, I conducted a pooled analysis of individual subject-level data from seven longitudinal training studies assessing hypertrophy outcomes in subjects with a wide range of body-fat levels. For more context and background on the analyses I ran, be sure to check out the footnote at the end of this article. Menno had a number of comments related to the analysis, which I am happy to address.
Menno states: “We have so many confounders you’d need major effect sizes and huge sample sizes to tease out any effects of less important variables like body-fat percentage.”
I think this is an important statement. When an effect is important and impactful, it’s usually not that hard to find. When trying to find evidence to support this p-ratio idea, it seems like we’re looking for a needle in a haystack. Football linemen are too genetically gifted, bodybuilders are too lean, and the paradigm we use to figure out the impact of virtually every training variable, dietary supplement, and recovery modality (a resistance training study without strict dietary control) simply won’t do. If this whole idea is really important enough to have a noteworthy impact on training and nutrition decisions, it should be easier to find than this.
In addition, this statement seems to imply that our analysis was inherently underpowered to detect small but practically relevant effects. On the contrary, our analysis did identify a statistically significant effect that wasn’t particularly enormous in magnitude. Baseline body-fat percentage was inversely associated with changes in fat mass (slope = -0.095, p = 0.001). This p-value was far below the significance threshold, and the slope indicates that someone who began the study with 25% body-fat would lose less than a kilogram more fat than someone who began the study with 15% body-fat. In other words, our meta-analysis was sufficiently powered to clearly identify a relatively small effect as statistically significant, with a p-value far lower than we’d “need” to establish statistical significance (implying that it was powered to also detect even smaller effects at higher, but still significant, p-values). The biggest factor preventing us from finding a significant relationship between baseline body-fat percentage and gains in lean mass is that, at least in this dataset, the two variables don’t seem to be reliably correlated to any meaningful degree.
Menno states: “As they acknowledge, they did not perform a systematic review, so there’s again potential for bias in study selection (though not consciously: I trust these guys absolutely).”
I appreciate Menno’s trust, but I think I understated our attempts to make this informal analysis as systematic as possible without threatening the feasibility of the project. Greg searched the literature by using Google’s dataset search tool, which indexes thousands of data repositories. He searched using several iterations of “resistance training hypertrophy” “resistance training lean mass” “resistance exercise hypertrophy” “strength training lean mass”, etc. and then screened every result. We didn’t catalog every step like we would if we planned on generating a PRISMA diagram to get the analysis published in a journal, but the only thing separating the search Greg performed and a “true” systematic search was a bit of cataloging and busy work. We included all of the studies that contained the information required for the analysis (body composition data before and after a resistance training intervention). If we were really leaning into this and trying to make it as systematic as possible, we probably would’ve emailed every author of every study that would have fit our criteria, but did not have publicly available data, to see if they’d be willing to share their data with us. This probably would have turned a five-day project into a five-year project, so we obviously passed on that. If you’re aware of any open datasets we missed (which is certainly possible, since datasets are harder to search for than studies; you can’t identify datasets that slipped through your search terms by following reference lists), we’d be happy to include them in our analysis. If we update the analysis with more data and the results change, then our conclusions will change in turn, but for now the data give us no strong justification for embracing the hypothesized relationship between body-fat percentage and p-ratios in lifters.
Menno states: “Lack of control for energy balance is a particularly major confounder considering their use of a ‘lean gains’ metric, defined as the change in fat-free mass minus the change in fat mass, rather than actual p-ratios, because it’s much easier to lose fat than to gain muscle. In studies without diet control, we typically see that some subjects lose bodyweight and fat, whereas others gain weight and probably more muscle. The ‘lean gains’ metric would majorly reward people ending up in energy deficit, even though we are actually mostly interested in muscle growth.”
In our article, we explained that we couldn’t use the p-ratio as the outcome variable (change in fat-free mass divided by change in body weight), because this can get a little messy with real-world data. For example, if someone “recomps” (gains muscle while losing weight), the simplistic p-ratio calculation becomes negative. Similarly, if someone loses muscle while gaining weight, they’d also have a negative p-ratio. This is unfavorable, because a negative p-ratio could simultaneously represent the best and worst possible outcomes. So, we made up our own “lean gains” metric to circumvent this issue (change in fat-free mass minus change in fat mass). We knew that some people would be dissatisfied or skeptical, so we also provided very straightforward analyses using fat-free mass and fat mass as outcome variables, just so nobody thought we were using some kind of nebulous, distorted outcome variable. As our original analysis showed, baseline body-fat was simply unrelated to changes in fat-free mass (slope = 0.0045, p = 0.797). Of course, we were also happy to look at more direct indices of hypertrophy; five of the studies included direct muscle thickness measurements, with some providing such values for multiple muscles. Instead of looking at hypertrophy in raw units, we scaled all of the values by their standard deviations (SDs) which allows us to get all of the measurements on fairly equal footing for a combined analysis. A quick visual assessment gives us little reason to believe that an individual’s baseline body-fat percentage was predictive of the magnitude of hypertrophy they achieved: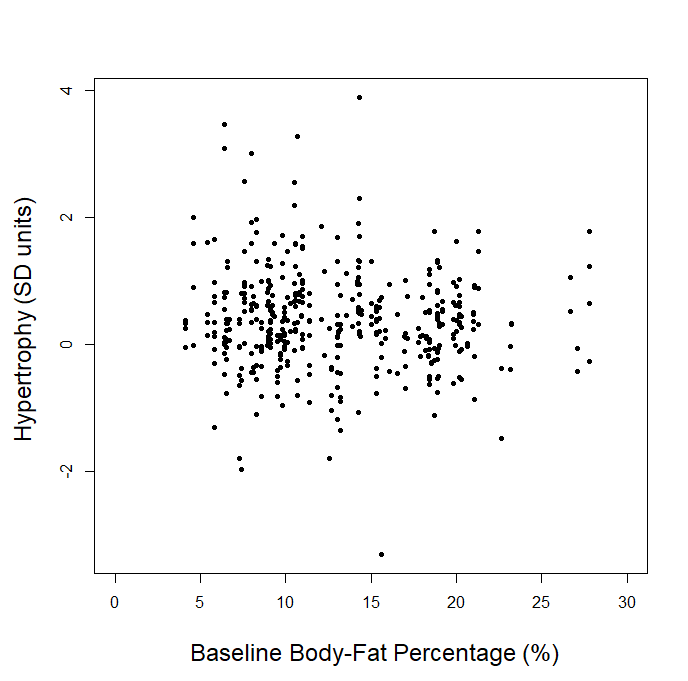
Visual inspection of the figure is informative, but we can re-analyze the data to see if baseline body-fat percentage influenced changes in direct hypertrophy measures while using random effects to account for the fact that multiple muscle measurements are nested within individual participants, and individual participants are nested within separate studies. When we run this analysis, we again observe non-significant findings (slope = -0.0009, p = 0.92), indicating that baseline body-fat percentage did not significantly influence direct measures of muscle hypertrophy in these studies. For the rest of this section, I’ll just focus on changes in fat-free mass as the main outcome of interest, as the remaining critiques specifically relate to lean mass accretion.
Menno states: “We’re looking at 1 variable in studies that were about something else, so you get major confounding effects. For example, did they gain more muscle because they were leaner or because they were in the group with the more effective training protocol?”
I was happy to alleviate this concern by accounting for “treatment group” in the analysis. So, instead of only accounting for the variance among studies, I also accounted for the variance among treatment groups, nested within the studies. With this alternate analysis, we still find a slope for changes in fat-free mass that is virtually zero (0.0039) with a p-value of 0.829 (decidedly not significant).
Menno states: “The analysis did not control for baseline muscle mass or training status, which obviously affects the rate of muscle growth. Lesser trained individuals may on average have more fat and less muscle and gain more muscle in a study, which would show up as a positive relation between body fat level and muscle growth.”
We could try to covary our way out of this situation; indeed, when we add baseline fat-free mass as a covariate, the relationship between baseline body-fat percentage and gains in fat-free mass remains negligible (slope = -0.0057, p = 0.760). However, an arguably simpler view is to actually control for training status by looking specifically at the studies with untrained participants. Aside from being the most likely to have higher inflammation levels and a pretty robust hypertrophy stimulus in response to 16-20 weeks of training, we can also be certain that individuals at all levels of body-fat have similar training status (none). This differs from studies that recruit trained subjects, as they often require that participants have a minimum level of training experience, but they rarely set a maximum, so training status isn’t necessarily restricted to a tight range. Whether you look at each of the studies with untrained participants individually, combine them, or combine them and analyze the male and female subjects separately, there is not a single analysis that gives us a practically meaningful slope for changes in fat-free mass or a p-value anywhere near the statistical significance threshold (all p-values were above 0.7).
Menno states: “They grouped men and women together. You cannot compare body fat percentages between men and women directly due to genetic differences in body fat storage patterns and essential fat levels.”
I anticipated that people would take issue with the combined analysis for numerous reasons, including this one. That’s why I included the results from within each independent study, in addition to providing the combined analysis with all studies lumped together. Looking at all of the studies independently, it certainly doesn’t appear that baseline body-fat level had a meaningful impact on lean gains in the studies sampling males, females, or a combination of both. Of course, I was also happy to stratify the combined analysis by sex to see if baseline body-fat percentage was predictive of fat-free mass gains in males or females. In males, the results were non-significant (slope = 0.011, p = 0.729). In females, the results were also non-significant (slope = 0.001, p = 0.962). Finally, when combining both into a single analysis that controls for sex, the results were still non-significant (slope = 0.002, p = 0.932). It seems safe to say that, at least within this dataset, sex was not meaningfully impacting the reported results. You can see the separate male and female data in the following figures:
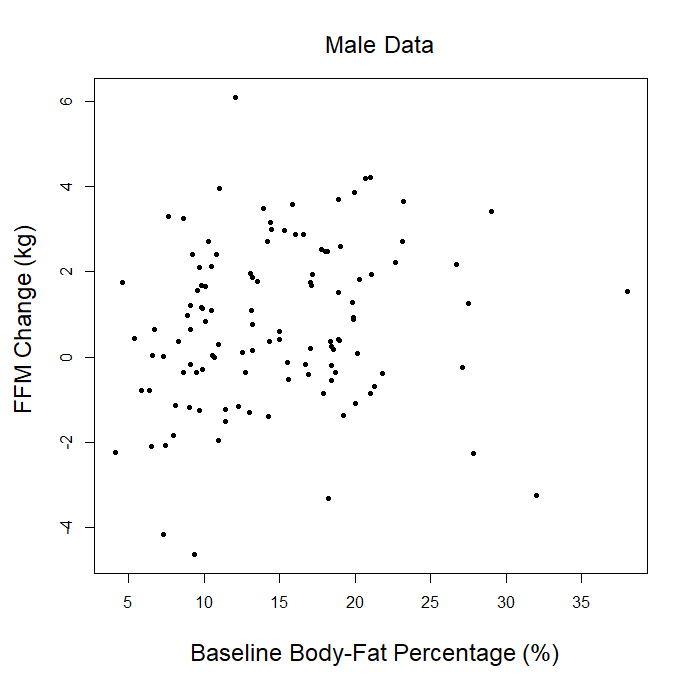
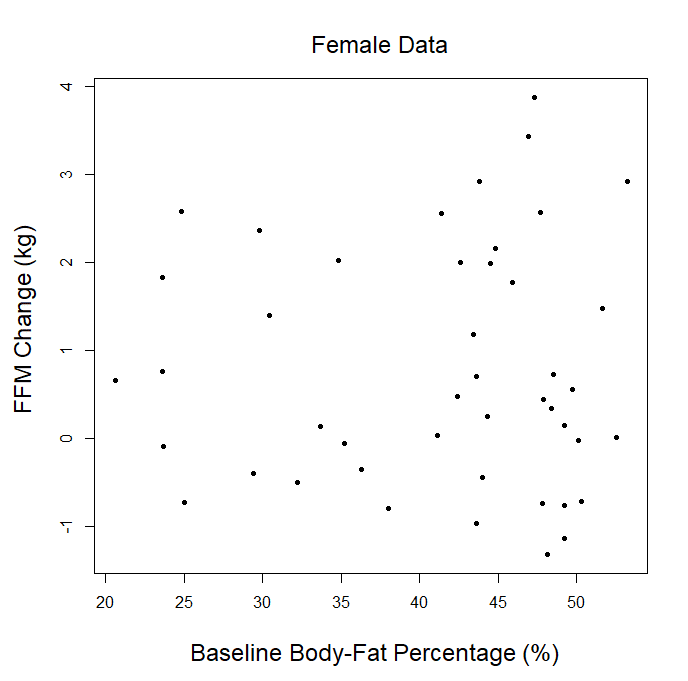
Menno states: “Most of these studies had no diet control. If people that eat more tend to have higher body fat levels and a greater likelihood of ending up in energy surplus, we can again get a positive relation between muscle growth and body fat percentage purely mediated by energy intake.”
In the original article, I provided the pooled results for our “lean gains” metric, but also provided the pooled results for changes in fat-free mass and for changes in fat mass. Given that baseline body-fat percentage had virtually no impact on changes in fat-free mass but was inversely (and significantly) associated with changes in fat mass, it should be clear that the proposed scenario (greater muscle growth due to a larger energy surplus) is not compatible with the data. Of course, we were also happy to calculate estimated energy balance for each subject based on changes in fat mass and fat-free mass (and the energy density of each; we used the same figures for energy density that Menno used in one of his previous articles) and re-analyze the data using energy balance as a covariate. After including this covariate, baseline body-fat was still not significantly predictive of changes in fat-free mass (slope = 0.004, p = 0.806).
Menno states: “Overall, I think a meta-analysis like this only has much promise if it systematically includes diet-controlled studies of a relatively homogenous population in energy surplus with considerable sample sizes. Then maybe we can observe a relation between the individuals’ p-ratios and their starting body fat level.”
We don’t need all of the studies to have large sample sizes, as a well-constructed meta-analysis can effectively incorporate data from studies of any size. Nonetheless, we all get to set our own requirements when it comes to what type of evidence we view as compelling or convincing. For this particular topic, this is where Menno sets his bar, and that choice is his to make. Curiously, his bar for the link between obesity-induced inflammation and hypertrophy is far lower, as is his bar for the link between body-fat and muscle protein balance. Further, his bar for legitimately questioning the relevance of this p-ratio idea is substantially higher than his bar for asserting that the idea is true, important enough to act upon, and beyond reproach. In evidence-based fitness, it generally works the other direction; we defer to the null hypothesis (you can add muscle pretty well in a very wide range of body-fat levels) until the evidence convinces us to adopt a less parsimonious hypothesis (we need to stay within a tight body-fat range because of inflammation, anabolic hormones, and whatever other mechanism is the flavor of the week).
In defense of our interpretation of the longitudinal data, let me lean on a metaphor. Let’s say there’s a brand new supplement on the market, and it makes sense from a mechanistic perspective (most do when they hit the market, to some degree). Based on this mechanism, we assume it’s going to help support hypertrophy in lifters. However, researchers finally start to do research on it; seven studies come out, with six showing no benefit, and one actually showing a statistically significant negative effect. Single studies often have questionable statistical power in the supplement literature, so someone combines the data from all seven studies to do a meta-analysis, but the meta-analysis still shows no significant benefit. One group of people concludes that the supplement isn’t worth the trouble; sure, it sounded good based on the proposed mechanism, but it just doesn’t seem to be panning out. Seems pretty reasonable to me, and that’s where we get with the vast majority of supplements that seem promising but end up falling flat.
A second group of people insists that the supplement does work, but we simply can’t observe it. The effect is so small that near-perfect conditions in huge studies would be required to actually identify a quantifiable effect, but you should still take it, and the entire evidence-based fitness community should embrace it. This second group asserts that the supplement is a robustly evidence-based intervention based on mechanistic studies that didn’t even involve ingesting the supplement, and even writes rebuttals when someone questions the efficacy of the product. No one catches flak for questioning the utility of a supplement with seven null or negative studies in a row; frankly, the less charitable skeptics would probably wonder if the adamant supplement proponents were getting kickbacks from the supplement company. When it comes to p-ratios, we’re expected to embrace this body-fat hypothesis as a robustly evidence-based principle that should guide our hypertrophy-related decision making based largely on inflammation studies that establish virtually no link between body-fat percentage and hypertrophy. We should bring our skepticism to the table when discussing new, flashy supplements that hit the market, but I don’t understand why we would leave our skepticism at the door when discussing p-ratios.
Admittedly, that’s not a perfect metaphor. For example, the studies in our p-ratio analysis weren’t explicitly designed to establish a relationship between body-fat percentage and hypertrophy outcomes. Nonetheless, they have all the information you’d need to do so. In addition, these studies are not literally the first seven studies in existence on a new topic; they were just the seven full datasets we could get our hands on. If people have empirical data (not mechanistic speculation) we missed that establishes a convincing link between high body-fat and impaired hypertrophy, we encourage people to share it, and we’ll happily revise our stance of body-fat and p-ratios. However, for now, the empirical data give us no strong basis for concluding that high body-fat levels impair p-ratios or blunt hypertrophy to an observable, let alone meaningful, degree in lifters.
General Note #1: Associations Upon Associations
Before wrapping up the article, it’s important to highlight that the argument in favor of body-fat messing up p-ratios by impairing hypertrophy is based on lots of transitive arguments linking indirect associations. For example, high body-fat is linked to inflammation, inflammation is linked to impaired hypertrophy, so high body-fat impairs hypertrophy. Unless both of those steps reflect a perfect 1:1 causal relationship, we can’t connect these dots with confidence. This type of misapplication of evidence underlies just about every fitness and nutrition myth that the evidence-based fitness world rolls their eyes at, so we ought to require stronger evidence before accepting a hypothesis.
General Note #2: The Effect of Training
We’ve alluded to this in various sections of this article, but the point is important enough that it bears repeating: resistance training has a positive impact on the mechanisms that are supposed to be linking high body-fat levels to impaired p-ratios and hypertrophy. Menno’s rebuttal discusses how high body-fat might have unfavorable effects on chronic inflammation, testosterone, cortisol, and recovery, and other proponents of the hypothesis have pinned the blame on impaired insulin sensitivity at higher body-fat levels. However, resistance training reverses a lot of these effects. The amount and consistency of the evidence and the magnitude of the effect varies from mechanism to mechanism, but there are studies suggesting that engaging in resistance training can favorably impact chronic inflammation, testosterone, cortisol, recovery, and insulin sensitivity. Admittedly, the testosterone and cortisol data are comparatively sparse and a bit inconsistent (likely due to contextual factors of each individual study), but the findings related to chronic inflammation, recovery, and insulin sensitivity are pretty solid. As such, we should be extremely cautious when extrapolating these mechanistic findings from untrained populations to trained populations, as training status influences the mechanisms themselves. This is why our original article placed so much emphasis on observations from people who were either resistance trained at baseline or undergoing a longitudinal training intervention, rather than purely looking at cross-sectional or acute evidence from totally untrained people. It’s totally possible that resistance training may be less effective in people with higher inflammation, lower testosterone levels, and higher cortisol levels if inflammation stays high, testosterone stays low, and cortisol stays high. However, once people start resistance training, those factors generally become more normal. The argument provided in the rebuttal to our p-ratio article fails to sufficiently consider this important point, as it leans on studies with very limited generalizability to people who are training regularly.
Caveats and Clarifications
In spite of the fact that lifting corrects or at least attenuates a lot of the metabolic and endocrine perturbations associated with obesity in sedentary individuals, it’s absolutely possible that there exists a level of adiposity that results in causal reductions in muscle anabolism. In our initial p-ratio article, we were responding to the very specific (and very popular) claim that resistance trained folks should specifically avoid high body-fat levels because p-ratios become unfavorable and hypertrophy becomes impaired at higher body-fat percentages. Some say that the advisable upper limit is 15%, whereas others say it’s as high as 25%. Some extend this logic to suggest that you can “game the system” by cutting to a lower body-fat to potentiate hypertrophy and enhance your subsequent muscle gains after the cut. Our analysis casts serious doubt on this concept within the body-fat ranges we observed (up to ~28% in trained males, up to ~38% in previously untrained males, and up to ~53% in previously untrained females). It’s also possible that high body-fat levels could hinder initial hypertrophy responses in totally untrained subjects, but given the effects of training on all of the purported mechanisms, such an effect should probably be attenuated within the first month or two. In the studies with previously untrained subjects that we analyzed, training interventions lasted 16-20 weeks, and baseline body-fat was unrelated to changes in fat-free mass.
It’s also worth noting that we analyzed data from studies that sampled fairly young participants (trained participants were under 40 years old and untrained participants were under 50 years old), so it’s possible high body-fat levels could potentially hinder hypertrophy in older people. We don’t have direct evidence to suggest that these effects truly are observed, we just have to acknowledge that our findings are specific to the body-fat and age ranges represented within the data we analyzed. Finally, our analysis of longitudinal data assessed the impact of body-fat by comparing the hypertrophy achieved by different people at different body-fat levels, rather than seeing how an individual’s ability to build muscle changes as that individual gains or loses body-fat percentage. That’s a limitation, but it’s a limitation shared by all of the human evidence used to support the hypothesized link between p-ratios and body-fat percentage. So, if you can reject the counter-evidence based on this limitation, you’ll probably have to throw out the supporting evidence as well. In addition, the limited longitudinal data that we do have suggests that p-ratios during weight regain are either impaired or unaffected by prior weight loss, so there’s no strong evidence to suggest that this limitation is masking an important advantage associated with cutting to get leaner before you bulk.
Nonetheless, our results indicate that for resistance trained people under 40 years old (or so), baseline body-fat doesn’t seem to impact gains in fat-free mass throughout the range of body-fat levels our data covers. Further, when we consider how the purported mechanisms linking body-fat to p-ratios are impacted by training, one might justifiably speculate that if elevated baseline body-fat does, in fact, negatively impact hypertrophy in untrained subjects, that effect would be attenuated after the first 4-6 weeks of regular training (such that its initial impact is undetectable after 16-20 weeks of training).
Claims We Aren’t Making
Now that we’ve clarified the claims we are making, it’s important to address a few claims that we aren’t making. Some people have responded to our initial article by noting that they don’t want to get over 15% or 20% body-fat for reasons unrelated to p-ratios. We think that’s totally fine, and our claims are agnostic to any individual’s goals. There are plenty of good reasons to stay leaner, whether they’re related to health, performance, or preference. We aren’t claiming that people need to get to high body-fat levels in order to make great gains, or that people necessarily should be intentionally bulking to extra-high body-fat levels. We’ve just heard people claiming that lifters are hindering their ability to make gains when their body-fat drifts beyond some arbitrary threshold, and we simply aimed to point out that there is insufficient evidence to make this claim.
Summary and Conclusions
The critique of my article indicates you should adjust your training and nutrition goals based on rodents without leptin receptors, 20-minute handgrip tasks during math class, fertility in obese couples, and varying degrees of inflammation among hospitalized, untrained elderly folks who are in bed 22 hours a day and completing a resistance training intervention that is literally less than a week long, while suggesting that comparing an offensive tackle to a tight end strains credulity. Moreover, the critique indicates you should lose body-fat on the basis of studies showing inflammation to be a mediator of hypertrophy, while the groups with high inflammation did not have higher body-fat. It speculates about how obesity impacts gains in resistance trained people, while showing no longitudinal, or even compelling cross-sectional evidence, of obesity independently impacting gains in response to resistance training. It suggests that obesity-induced inflammation directly impairs muscle protein balance, directly blunts lean mass gains in response to resistance training, and tanks testosterone levels to an extent that the accretion or maintenance of large amounts of lean mass is untenable, yet concedes that high adiposity is a prerequisite for anyone wishing to achieve their maximal level of muscularity. When pondering whether or not leaner people would inherently make better gains in response to the same training intervention, the critique asks you to dismiss subject-level human data observing longitudinal responses to standardized resistance training programs in people covering an extremely wide range of body-fat levels.
So, I once again ask the question I was asking before I wrote the original article: Where is the evidence? Where are the documented instances of people who are struggling to gain lean mass because their body-fat was too high when they started the training intervention? Why are we trying this hard to lean on mice without leptin receptors and six-day resistance training interventions in hospitalized elderly people when there are infinitely more generalizable sources of data available?
Notably, my question is not “can you conceive of a mechanism that might link obesity to impaired gains?” While I was cautious to note the limitations of my indirect evidence, it certainly fails to show a negative independent impact of high body-fat that is substantial enough to care about. While I was cautious to note the limitations of our combined analysis of subject-level data, it gives us no reason to believe that when exposed to the same intervention, leaner people should expect better gains than people with higher body-fat at baseline. In addition, I was happy to explore the various speculative confounders raised in the critique of my article, with none of these re-analyses bringing us meaningfully closer to evidence of high body-fat level inhibiting gains.
As I highlighted in the original article, it is easy to extrapolate and theorize based on plausible mechanisms, but this falls short of constituting compelling evidence. So, while my original article focused on the flimsy case of insulin-based speculation, shifting the focus toward inflammation-based speculation doesn’t really take us any closer to empirical evidence connecting the dots between high body-fat and impaired lean mass gains. Of the hundreds and hundreds of studies that get published each year, several observe body composition changes associated with resistance training, and each subject in each study has a body-fat percentage. Nonetheless, it seems hard to find empirical evidence supporting this relationship between body-fat and hypertrophy potential. Thus, after assessing the arguments provided in a full article and a full rebuttal, proponents of the concept find themselves in a familiar spot: heavy on mechanistic speculation, heavy on theorizing, but light on data.
Perhaps the best example reinforcing this vagueness arises when you ask a few proponents of this p-ratio hypothesis to put a number on it – that is, at what body-fat percentage do p-ratios get out of whack and threaten hypertrophy responses? In my experience, the number provided (for males) may be as low as 15% body-fat, or as high as 25%, depending on who you talk to. For an idea that is often presented as being evidence-based, one might wonder how the base of evidence has led to such markedly different estimates. Surely the two camps can dig into the evidence and sort out the discrepancy, provided that the estimates are not based on a hunch or undocumented anecdotes. Some people are quite forthcoming, and admit that their belief in the concept (and the upper-limit body-fat percentage they recommend) isn’t actually based on particularly relevant evidence, and that is totally fine. I take no issue with people turning hunches and anecdotes into practical recommendations, as long as they don’t claim that the recommendation is based on robust scientific evidence. I don’t have enough evidence to assert that this whole p-ratio hypothesis cannot exist, my article simply indicates that I can’t find any convincing evidence to support it, and I can find some indirect evidence that seems to contradict it. My article can’t be used to permanently and conclusively “debunk” the concept, but at the very least, it should encourage people to discuss the concept in a way that effectively conveys the tremendous amount of speculation and surprising lack of support that the concept rests upon.
Footnote: How the Models Work
In our various analyses, we’re trying to see if baseline body-fat percentage is predictive of our outcome variables of interest (changes in fat mass, changes in fat-free mass, or changes in our “lean gains” metric over the course of a study). Another way to phrase that is we’re trying to determine the degree to which baseline body-fat percentage explains the variance in our outcome variable in the model. If the p-value is less than 0.05, we’d conclude that baseline body-fat is significantly predictive of the outcome, and we’d use the slope of the regression line to help us interpret that relationship. For example, imagine that we’re trying to determine if baseline body-fat percentage is predictive of changes in fat mass, and we get a slope of -0.09kg. This means that for every one-unit increase in baseline body-fat percentage, we’d expect an individual to lose an extra 0.09kg of fat mass over the course of their study. If we compared two people whose baseline body-fat percentage differed by 10 percentage points (that is, 15% body-fat versus 25%), then we’d expect the person who started at 25% body-fat to lose an extra 0.9kg of fat over the course of their study when compared to someone who started at 15% body-fat.
In our analyses, we combined the results from multiple studies into big, pooled regression models. If we jam data from multiple distinct studies into a standard regression model without making any adjustments, we have to assume that 0% of the variance in the outcome variable (whichever one we’re analyzing at any given time) is attributable to factors that differ among studies. In other words, we’d have to assume that the studies are perfectly homogeneous, such that two data points from the same study are no more similar to each other than they are to the data points from other studies, and that’s not a good assumption. So, we introduced a random intercept for “study”; this models the variance in our outcome variable that is attributable to differences among studies, and effectively remedies our issue of certain data points being more related to each other than they are to other data points.
In this rebuttal, we made things a little more complicated, but not by much. We mentioned “controlling” for certain variables. That just means we added them to the regression model as a “covariate” to see how well baseline body-fat percentage explained the variance in our outcome variable after controlling for the covariate. We also did one analysis where we added random intercepts for “study group” in addition to “study.” In this scenario, we’re modeling the variance among different treatment groups within a given study, while also modeling the variance among different studies within the dataset. This allowed us to minimize any concern that our results were being drastically distorted by wildly divergent effects that varied from study group to study group. Similarly, this rebuttal included a model for direct measures of hypertrophy in which random intercepts were included for “participant” and “study,” because some of the studies reported multiple different muscle hypertrophy measurements per subject. So, the models are basically just regular old regression, with a few extra bells and whistles to account for stuff that needs to be accounted for. It may seem weird that we are doing this with subject-level data from individual participants, and you may have concerns about the concept of combining participants from different studies with slightly different interventions and sample characteristics. However, you’re probably quite familiar (and comfortable) with meta-analyses, which do the same thing with aggregated sample-level data (that is, they combine the overall summary data from several different studies rather than the individual-level data from several different studies). Rest assured, if every meta-analyst actually could use the individual-level data, they probably would, but it’s rarely feasible due to restricted access to full datasets. If publicly available datasets become more common, you might start to see more analyses like the ones we have presented in this article.


